







En definitiva, la Reacción en Cadena de la Polimerasa (PCR) es una técnica de laboratorio que se utiliza para realizar millones de copias de un segmento de ADN específico. Los científicos a menudo necesitan estudiar el ADN, pero las muestras biológicas (como una gota de sangre o un hisopo de una mejilla) generalmente contienen una cantidad muy pequeña de material genético, demasiado poco para verla o analizarla directamente. La PCR resuelve este problema de “aguja en un pajar” amplificando el ADN hasta que haya una cantidad lo suficientemente grande con la que trabajar.
Cómo funciona la PCR: el ciclo de tres pasos
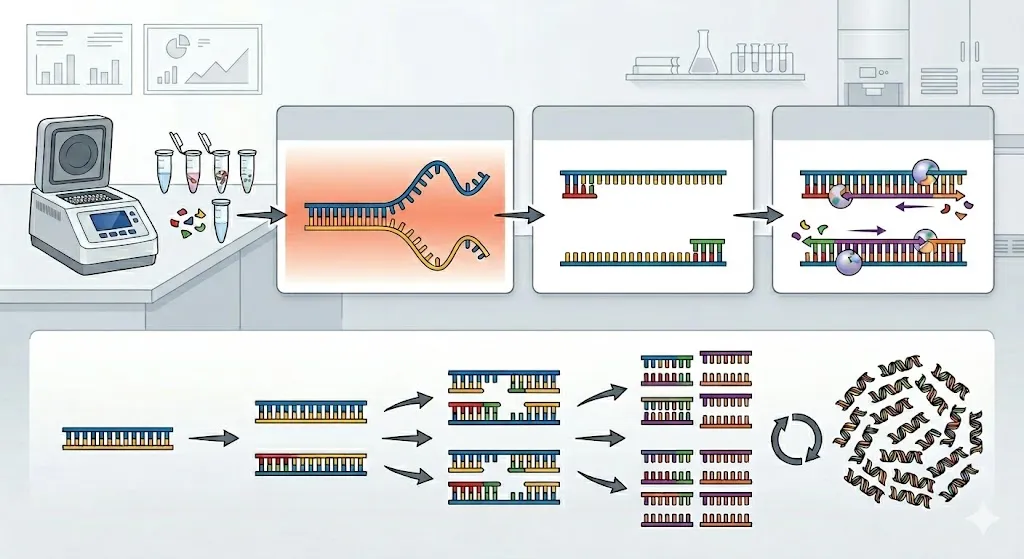
La PCR no requiere maquinaria compleja para “cortar y pegar” ADN; en cambio, utiliza cambios de temperatura para controlar la reacción. El proceso ocurre dentro de un termociclador y se repite aproximadamente de 25 a 40 veces.
- Desnaturalización (El “Descomprimir”)
La mezcla de reacción se calienta a aproximadamente 95°C. A esta alta temperatura, los enlaces de hidrógeno que mantienen juntas las dos hebras de la doble hélice del ADN se rompen, lo que hace que el ADN se separe en dos hebras individuales.
- Recocido (El “Prime”)
La temperatura se reduce a entre 50°C y 65°C. Esto permite que fragmentos cortos de ADN creados a medida, llamados cebadores, se unan (hibriden) a secuencias objetivo específicas en el ADN monocatenario. Estos cebadores actúan como “marcadores” y le indican a la enzima exactamente dónde comenzar a copiar. 3. Extensión (La “Construcción”)
La temperatura se eleva a aproximadamente 72°C. Una enzima llamada Taq polimerasa (una ADN polimerasa termoestable) se agarra a los cebadores y comienza a agregar nucleótidos a la cadena. Construye una nueva cadena complementaria de ADN, duplicando efectivamente la cantidad de ADN objetivo.
Protocolo práctico de PCR y resolución de problemas
Configure una reacción de 25 l en hielo: 12,5 l de mezcla maestra de PCR 2 × (que contiene polimerasa Taq, dNTP, MgCl2 y tampón), 0,5 l de cada uno de cebadores directos e inversos (stock de 10 m, concentración final 0,2 m), 1 a 100 ng de ADN plantilla y agua libre de nucleasas hasta 25 l. Diseñe cebadores con 18 a 24 nucleótidos, entre 40 y 60 % de contenido de GC y una temperatura de fusión de 55 a 65 °C. La temperatura de recocido óptima (Ta) es de 3 a 5 °C por debajo de la Tm del cebador; ejecute un gradiente de 50 °C a 65 °C para determinar la mejor Ta. Evite cebadores con abrazaderas 3’ GC de más de 3 bases, ejecuciones del mismo nucleótido de más de 4 o estructuras de horquilla previstas. Amplifique usando una desnaturalización inicial a 95 °C durante 3 minutos, seguida de 30 a 35 ciclos de 95 °C durante 30 segundos, Ta durante 30 segundos y 72 °C durante 30 a 60 segundos por kb de objetivo, con una extensión final a 72 °C durante 5 minutos. Si no se obtiene ningún producto, aumente la cantidad de plantilla, reduzca Ta entre 2 y 5 °C o agregue DMSO (2 a 5 %) para plantillas ricas en GC. Para bandas no específicas, aumente Ta, reduzca MgCl2 a 1,5 mM o disminuya la concentración del cebador. Si se corre, reduzca el número de ciclos o la cantidad de plantilla. Analice 5 µL de producto mediante electroforesis en gel de agarosa con una escalera de ADN para confirmar el tamaño.
Aplicación del mundo real
Al genotipar un SNP humano (rs1800497), la PCR con cebadores específicos de alelo amplifica un producto de 320 pb del ADN genómico extraído de hisopos bucales. Un gradiente de temperatura de recocido identifica 60°C como óptimo. Después de 35 ciclos, la electroforesis en gel muestra una única banda limpia en todas las muestras. El producto es secuenciado por Sanger para confirmar el polimorfismo Taq1A, lo que demuestra la importancia del diseño de cebadores y la optimización térmica para un genotipado reproducible.
recurso: Calculadora Lab Lexicon Oligo Tm